




Spettroscopie computazionali per adsorbati e nanoparticelle
This research line focus on the characterization of molecular based materials by spectroscopic techniques for electronic excited states in the low and high energy region of the spectrum. This kind of study represents one of the most fruitful areas of synergetic interplay between experiment and theory and in fact strong collaborations with researchers of the GAS-PHASE and ALOISA beamlines at Elettra synchrotron radiation laboratory are active. The more general theoretical activity concerns the calculations of electronic excited states by means of computational methods within the realm of density functional theory (DFT) and its extension to the time dependent approach (TDDFT). Two main tasks are pursued:
1. Study of the core excited states in molecules adsorbed on surfaces.
2. Study of the valence optical properties in metal particles
1. The Near Edge X-ray Absorption Fine Structure spectroscopy (NEXAFS) of molecules adsorbed on surfaces is widely used to investigate the orientation and geometry of the molecular adsorbate as well as the extent of the adsorbate-substrate interaction. The simulation of NEXAFS spectra of these systems represents a computational challenge for both a proper modelling of the adsorbate/surface system as well as for its size which can prevent the application of very accurate theoretical methods. We calculate the NEXAFS spectra in terms of excitation energies and oscillator strength values calculated at DFT or TDDFT level employing a cluster approach to mimic the adsorbate systems. Finite size cluster model for surface adsorption has proven very efficient and accurate to simulate NEXAFS spectra due to the localized nature of the core excitation process. Total spectra as well as angle resolved spectra can be simulated. Previous optimization of the surface and of the molecules adsorbed on it is performed by a periodic slab methodology in the frame of density functional theory (DFT)
Some recent studies on primary amines on Au(111) surface in collaboration with researchers of the ALOISA beamline of the Elettra Synchrotron in Trieste are briefly presented.
Computational Study of Amino Mediated Molecular Interaction Evidenced in N 1s NEXAFS: 1,4-Diaminobenzene on Au (111)
G. Balducci, M. Romeo, M. Stener, G. Fronzoni*, D. Cvetko*, A. Cossaro, M. Dell’Angela, G. Kladnik, L. Venkataraman, A. Morgante
J. Phys. Chem. C (2015) 119, 1988-1995
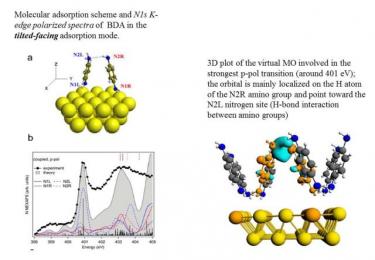
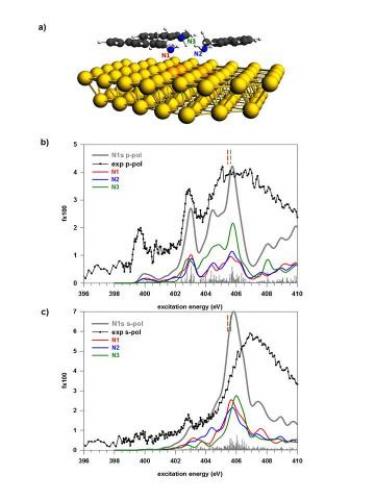
The primary amines can interact with neighbor molecules or with the substrate via weak bonds involving the electron lone pair of their amino functional group. Near edge x-ray absorption spectra (NEXAFS) on the N1s edge show that the structure of the empty molecular orbitals localized on the nitrogen atom is very sensitive to these interactions. In this study the NEXAFS spectra are simulated for the 1,4-benzenediamine (BDA) molecule in its free, crystalline and monolayer on Au(111) forms and compared with the experimental data.
The chemistry of the methylamine termination at a gold surface:
from auto-recognition to condensation.
In preparation
The theoretical simulation of the N1s spectra concerns the monolayer flat phase where both the amino-gold and the intermolecular amino-amino interactions can be analyzed in detail.
The configuration reported in the figure explores the issue of the interplay between the amino-gold and the amino-amino interactions through hydrogen bond by considering a model with three NMA adsorbed molecules. The experimental dichroic effects are well reproduced by the calculations.
| |
|
Gruppo di ricerca
| Gruppo Spettroscopie computazionali |